Protax
Med detta verktyg kan du bestämma vilken som helst DNA-sekvens från en finsk insekt eller spindel, bara sekvensen härrör från den vanliga DNA-streckkodregionen, COI, och matas in i FASTA-format. Programmet kan inte identifiera andra taxa än insekter och spindlar.
Klistra in DNA-sekvensdata (i FASTA-format) i textfältet nedan eller välj filen med filväljaren.
- Textfält
- Filväljare
.
Läs mer om Protax och om hur du tolkar resultaten
Instruktioner
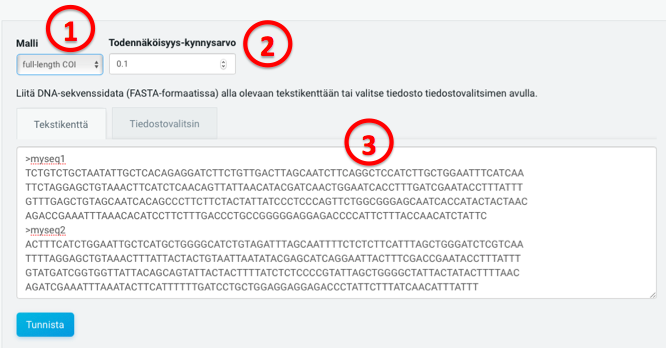
(1) Välj mellan en modell baserad på hela streckkodsregionen eller på Leray-regionen.
Utgående från detta val tillämpar identifikationsverktyget antingen en modell baserad på hela Folmer-regionen (658 bp) eller på Leray-regionen (313 bp). Om du inte är säker på att alla inmatade FASTA-sekvenser representerar Leray-regionen skall du välja modellen för hela streckkodsregionen. Denna modell fungerar även för Leray-sekvenser, även om den Leray-specifika modellen är snabbare.
Du kan prova med olika exempeldata: 10 sekvenser, 100 sekvenser eller 6486 sekvenser.
(2) Välj en sannolikhet (mellan 0 och 1) och ett minimumvärde för hur mycket sekvenserna skall överlappa (mellan 1 och 500)
För varje inmatad sekvens kommer programmet att inkludera en sannolikhet för varje taxonomisk placering som överskrider detta tröskelvärde. Således kan valet av ett lågt tröskelvärde resultera i en mycket stor utfil med väldigt många alternativa taxa och tillhörande sannolikheter.
Minimumvärdet för hur mycket sekvenserna skall överlappa med varandra anger kortaste tillåtna överlapp mellan en inmatad DNA-sekvens och en referenssekvens. Genom att specificera detta värde kan användaren reducera inverkan av speciellt korta och därmed oinformativa sekvenser på sina resultat. Den verkliga längden på en sekvenslinjering avgörs nämligen av antalet sekvenspositioner för vilka ingendera sekvensen innehåller någon lucka – även om det parvisa avståndet mellan två sekvenser nominellt beräknas utgående från deras globala linjering. Eftersom vissa av våra referenssekvenser saknar partier i början på sekvensen, så kan den verkliga linjeringen bli mycket kort om den inmatade sekvensen begränsar sig till just precis början på COI-genen. Genom att specificera ett lägsta tröskelvärde för längden på sekvenslinjeringen minskar vi effekterna av sådana brister.
(3) Ange 1-2000 COI-sekvenser i ett FASTA-format; du kan antingen kopiera och klistra in dem i textfältet eller specificera ett filnamn i filväljaren.
Indata måste representera samma DNA-sträng som referenssekvenserna. Med andra ord måste DNA sekvensen vara avläst i den riktning som anges av den framåtriktade primersekvensen vid standard COI PCR. Maximal filstorlek är 4 MB, vilket motsvarar 6000-7000 COI-sekvenser.
Klicka på Identifiera så får du en zip -fil som innehåller två filer:
En textfil som anger sannolikheten för varje taxonomisk placering på varje nivå i taxonomin. Denna klassificering görs separat för varje inmatad sekvens.
En HTML -fil som visar resultaten som ett Krona-diagram.
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No 856506).