Ett bestämningsverktyg för DNA-sekvenser från finska leddjur
Denna sida beskriver bakgrunden till det nya Protax-verktyg som nu tillåter bestämningen av DNA-sekvenser från finska insekter och spindlar.
Bakgrund
Bestämningsverktyget använder sig av den samlade informationen i alla de insektsekvenser som fogats till det nationella streckkodbiblioteket, FinBOL. Detta gör det möjligt att avgöra med vilken sannolikhet ifrågavarande sekvens tillhör ett visst taxon på en viss taxonomiska nivå (klass, ordning, familj, underfamilj, tribus, släkt och art) – eller ett tidigare okänt taxon på samma nivå. Den senare sannolikheten bör uttryckligen tolkas som "ett taxon som saknas ur referensbiblioteket" (inklusive taxa helt andra än insekter).
FinBOL-referensbibliotekets omfattning framgår ur Roslin et al. 2021. Eftersom verktyget är intränat på informationen i en bestämd genregionen så måste användaren ange vilka primersekvenser hen använt
Hur man tolkar resultaten
För varje inmatad sekvens anger programmets utskrift sannolikheten för varje alternativ taxonomiska placering. Därför sträcker sig resultaten för varje sekvens vanligtvis över flera rader, varvid varje rad innehåller sannolikheten för taxonomisk placering i just detta taxon. En given sekvens kan vanligtvis ges flera alternativa taxa på samma taxonomiska nivå – och hur många, det beror det på vilket tröskelvärde för sannolikhet som användaren specificerat. Det finns fem kolumner på varje rad:
seqid taxonomic_level finbif_taxon_code taxon_name probability
Seqid är sekvensens id – den som angivits på rubrikraden för varje sekvens i FASTA-filen.
Det finns sju taxonomiska nivåer: 1) klass, 2) ordning, 3) familj, 4) underfamilj, 5) tribus, 6) släkte och 7) art. De taxonomiska namnen följer checklistan för finska arter och sannolikheten är sannolikheten för att sekvensen ifråga tillhör ifrågavarande taxon.
Nedan följer tre exempel på hur man ska tolka innehållet i utdata (se även anteckningen i slutet av detta dokument).
Exampel 1: Tillförlitlig identifiering ned till artnivå.
Sannolikheten för en bestämd taxonomisk placering minskar när man går från klassnivå till artnivå, eftersom det behövs mer information för att knyta en sekvens till en lägre taxonomisk nivå. I detta exempel är dock sannolikheten för att sekvensen kan knytas till en bestämd art hög. Därför är det mycket troligt att sekvensen 'GMFIH293-12' tillhör arten Macaria notata.
Example 2: Tillförlitlig identifiering till familjenivå

I det här exemplet uppnår vi bara en bestämning på underfamiljnivå, och här förs taxonet till "unk". Unk står för okänt (unknown), dvs taxa som saknar referenssekvenser. Denna kategori omfattar både sådana kända taxa som saknar sekvenser och tidigare okända taxa. Tolkningen av resultatet blir därmed att sekvensen troligen tillhör familjen Braconidae, men under Braconidae tillhör ett taxon för vilket vi saknar referenssekvenser.Obs! För okända taxa sakans taxonkoder i FinBIF-taxonkod, och de ersätts därför med NA (för Not Available, ej tillgängliga).
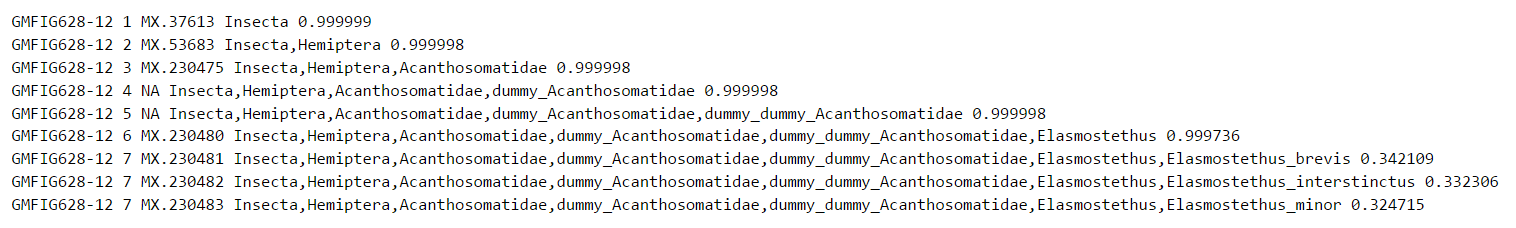
Exempel 3: Identiska streckkoder
I det här exemplet uppnår vi en hög sannolikhet ner till släktnivå, men på artnivå fördelas sannolikheten jämnt mellan tre olika arter. I detta speciella fall har de tre arterna faktiskt identiska streckkodssekvenser. Därför räcker inte den vanliga COI-sekvensen för att avgöra vilken art sekvensen kommer ifrån. Tolkningen av resultatet blir då att sekvensen sannolikt representerar släktet Elasmostethus, och att den inom detta släkte tillhör en av arterna E. brevis, E. interstinctus eller E. minor. För att avgöra vilken av dessa arter det rör sig om skulle vi därmed behöva mera information. Om t.ex. någon eller några av dessa arter saknas från den lokal där sekvensen samlats in så begränsar det urvalet av möjliga arter.
Detta exempel visar också på ett namngivningsproblem orelaterat till de identiska streckkoderna: På nivå 4 och 5 i taxonomin ovan uppträder ordet "dummy". Detta beror på att den taxonomi som använts för att träna Protax saknar namn på taxonomiska nivåer mellan familjen Acanthosomatidae och släktet Elasmostethus; här innehåller taxonomin i FinBIF inga underfamiljer eller tribus Eftersom Protax utgår ifrån att varje rutt från rot till art i taxonomin har samma antal nivåer (sju), så har vi ersatt de mellannivåer som saknats med namn som börjar med "dummy". Detta tekniska hjälpmedel påverkar inte utfallet, dvs sekvensen är troligtvis associerad med ett släkte Elasmostethus.
Obs: Eftersom FinBIF saknar taxonkoder för taxonnamn med efterleden ”dummy" ser vi koden ”NA” (Not Available) i den tredje kolumnen.
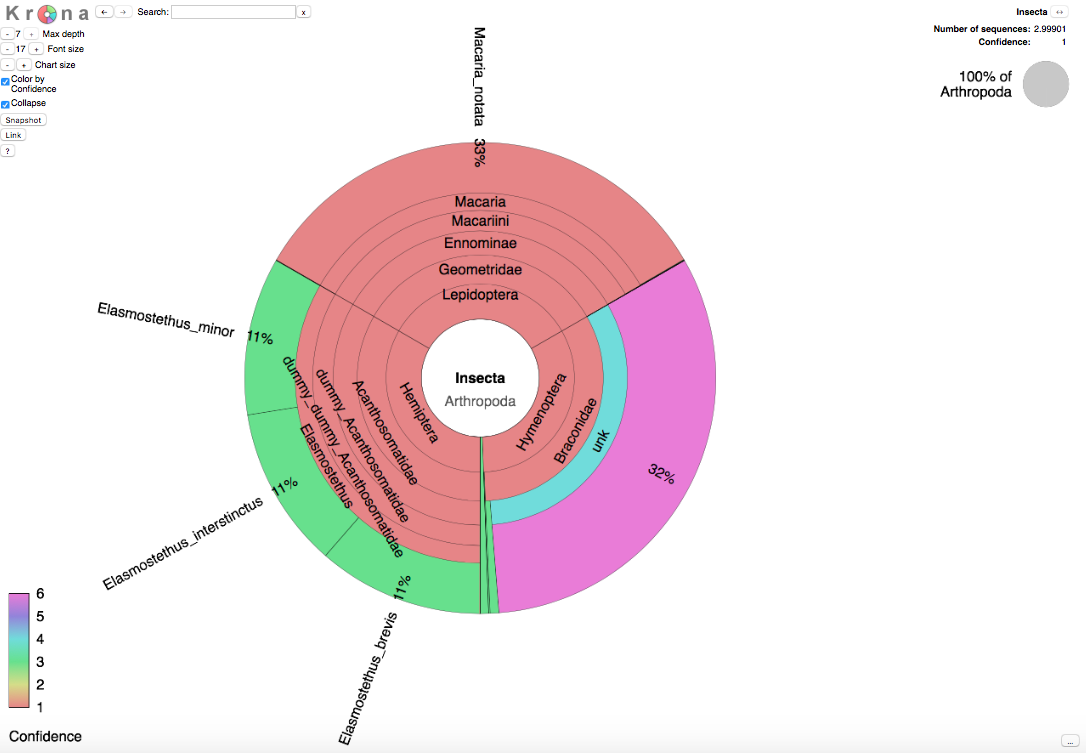
Ett taxonomiskt tårtdiagram
Förutom textfilen med sannolikheten för olika taxonomiska placeringar för varje inmatad sekvens, så innehåller utskriften också en html-fil där resultaten framställs i form av ett interaktivt taxonomiskt tårtdiagram. Detta diagram har producerats med hjälp av verktyget Krona . Detta verktyg tillåter användaren att undersöka resultaten på alla taxonomiska nivåer.
Genom att klicka på "Color by confidence" till vänster på skärmen kan du färglägga varje taxon med den osäkerhet som kvarstår vid bestämningen. Kategorier 1-3 motsvarar taxa för vilka referenssekvenser funnits tillgängliga och kategorier 4-6 motsvarar taxa för vilka referenssekvenser saknas.
- (röd). Mer än hälften av sekvenserna i detta taxon uppnår en sannolikhet som överstiger 0,9
- (gul). Minst en av sekvenserna i detta taxon uppnår en sannolikhet över 0,9
- (grön). Ingen sekvens i detta taxon uppnår en sannolikhet som överstiger 0,9
- (turkos). Mer än hälften av sekvenserna uppnår en sannolikhet över 0,9
- (blå). Minst en av sekvenserna uppnår en sannolikhet över 0,9
- (magenta eller anilinrött). Ingen sekvens uppnår en sannolikhet över 0,9Storleken på varje taxon visar det antal sekvenser som tillskrivits det. Detta antal är summan av alla sannolikheter för denna taxonomiska placering, summerad över alla inmatade sekvenser.Bilden nedan visar det Krona-diagram som uppstår när man matar in exempelsekvenserna ovan.
Hur fungerar metoden?
För taxonomisk placering behövs en taxonomi, en uppsättning referenssekvenser och en modell för att konvertera sekvensernas (dis)similaritet till taxonomiska sannolikheter. Vi använder den probabilistiska taxonomiska klassificeraren PROTAX. För varje inmatad sekvens genomför vi följande steg:
- Linjera sekvensen mot referenssekvenserna med hjälp av en dold Markov modell (hidden Markov model)
- Beräkna p-avstånd mellan den inmatade sekvensen och alla referenssekvenser
- Omvandla p-avstånd till sannolikheter med hjälp av en multinomial regressionsmodell
- Skriv ut alla taxa och sannolikheter som överstiger det gränsvärde som angivits av användaren
P-avstånd är antalet avvikande nukleotider dividerat med längden på linjeringen. Här beaktas endast de positioner som kännetecknas av ett A, C, G eller T i både den inmatade sekvensen och referenssekvensen, dvs luckor och N-baser utesluts. För mer information om metoden, se Roslin et al. 2021. För mer information om PROTAX, se referenser Somervuo et al. 2016a och Somervuo et al. 2016b.
OBS: verktyget identifierar bara insekter och spindlar
Verktyget har tränats in på finska insekter. Majoriteten av referenssekvenserna representerar insekter och en minoritet Arachnida. Därför har verktyget ingen som helst uppfattning om COI-sekvenser från andra taxa. Om användaren matar in COI-sekvenser för något annat taxon än Insecta eller Arachnida kommer verktyget följaktligen inte att kunna ange deras ursprung. Isället kommer sekvenserna att förknippas med en hög sannolikhet för ”Insecta, unk” och med låga sannolikheter för bestämda ordningar eller lägre nivåer. För att minska beräkningsbördan vid klassificering av massdata kan användaren därför göra klokt i att först att använda någon grövre klassificerare för att filtrera bort sekvenser av andra än leddjur innan hen tillämpar Protax-verktyget på de återstående sekvenserna.
Referenser
- Roslin, T., [95 medförfattare] & Mutanen, M. 2021. A molecular-based identification resource for the arthropods of Finland. Molecular Ecology Resources https://doi.org/10.1111/1755-0998.13510.
- Somervuo P, Koskela S, Pennanen J, Nilsson HR, and Ovaskainen O. Unbiased probabilistic taxonomic classification for DNA barcoding. Bioinformatics. 2016 Oct 1;32(19):2920-7. doi: 10.1093/bioinformatics/btw346
- Somervuo P, Yu DW, Xu CCY, Ji Y, Hultman J, Wirta H, and Ovaskainen O. Quantifying uncertainty of taxonomic placement in DNA barcoding and metabarcoding. Methods in Ecology and Evolution. 2017 8,(4): 398-407. doi: 10.1111/2041-210X.12721